
本文根据“rAAV研究先锋”Richard Jude Samulski 近期发表在Molecular Therapy杂志(IF=12.1)上的一篇综述进行整理,该综述系统回顾了 AAV 技术 40 年的演进,并展望人工智能驱动的衣壳工程、精准递送及rAAV药物的临床试验和商业化等未来趋势[1],是理解 AAV 行业过去与未来的权威读物。

我们将分几期对该综述进行详细介绍,本期我们重点介绍AAV药物临床试验进展与现实挑战。文章有点长,但是真的值得你收藏。
一、AAV基因治疗临床试验进展盘点
AAV首次临床试验始于1995年,由Terry Flotte博士进行的CFTR递送研究。根据最新数据,截至2025年1月27日,全球已登记AAV相关临床试验343项(2022年为255项),增长近35%(图1)。

图1 AAV 的临床试验数量随时间变化的趋势(统计截止至 2025 年 1 月 27 日)。
不同阶段的临床试验数量如下:
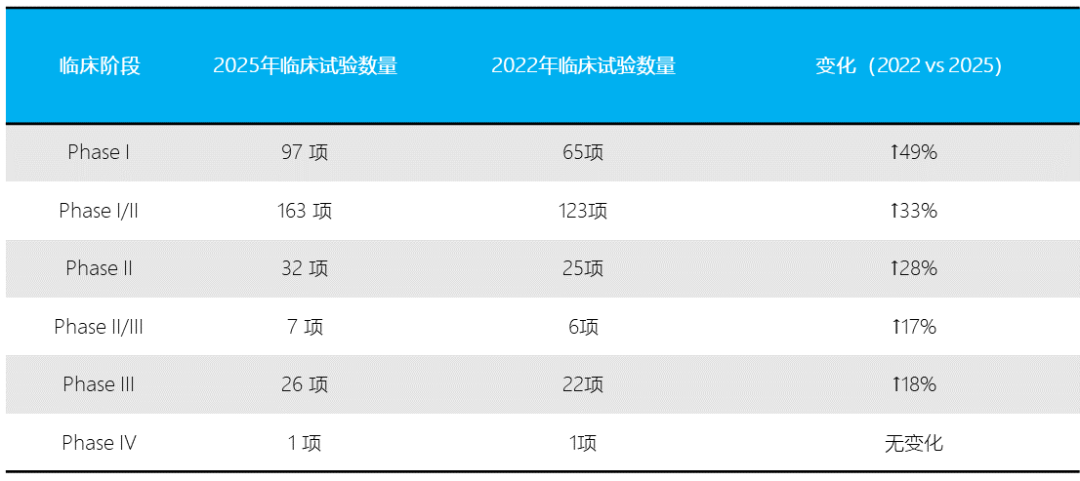
尽管I/II期数量不断增长,但进入III期的比例仍然有限(图2A),提示AAV载体在推进至后期阶段时仍面临技术与监管挑战。

图2 AAV临床试验概览。(A) 不同临床阶段的AAV临床试验分布情况;(B) 按照适应症类型对AAV临床试验进行分类统计;(C) 根据AAV载体靶向的组织/器官类型对临床试验进行分布分析;(D)不同给药途径的使用比例统计。
2. 适应症的选择
虽然“一基因一疾病”的治疗逻辑看似完美,但现实远比设想复杂。截至目前全球罕见病总数约 7,000~8,000 种,当前AAV试验仅覆盖 约80种疾病,占比 <1%。临床试验集中于 血友病A/B、肌营养不良、视网膜色素变性、湿性AMD、帕金森、Leber先天性黑蒙等。其中,湿性/干性AMD、心力衰竭、帕金森等并非罕见病,提示AAV也在逐步扩展其治疗范围。
目前AAV基因治疗的研发重点仍集中在眼科领域,其次是中枢神经系统(CNS)和肝脏,三者分别占据26%、21%和18%的临床试验份额(见图2C)。这一分布与Shen等人两年前的统计结果基本一致,说明在适应症选择策略上,过去两年并未出现重大调整。这也提示我们,目前AAV载体在临床应用中所面临的挑战(如肝毒性)很可能具有器官特异性,只有在不同器官体系中积累足够研究数据并进行横向比较后,才能更全面理解其机制。
4. 递送途径:单一路径为主,双途径策略逐渐涌现
在临床试验中,AAV递送采用了多种给药方式,如图2D所示。其中,与靶向组织相对应,血管内注射仍是最常用的途径,其次是用于眼部适应症的三种主要给药方式(即视网膜下、玻璃体内、脉络膜上注射),以及用于中枢神经系统递送的三种方式(脑室内注射ICV、大脑实质内注射、鞘内注射IT)。
5. 蛋白环境对递送效果有深远影响
随着我们对AAV与宿主细胞环境中蛋白质(如血清、脑脊液、玻璃体液)相互作用机制的深入了解,研究人员开始意识到,不同给药途径的效果可能受到各组织微环境中特异性蛋白的增强或抑制影响。
值得注意的是,已有4项临床试验采用“双途径联合递送”策略,包括血管内注射联合ICV、血管内注射联合IT、ICV联合玻璃体注射,以及实质内注射联合ICV。这类多路径递送策略并未在饼图中单独展示,但其复杂性显著增加,往往需要为每种途径设计不同的衣壳,导致生产、毒理研究等环节的成本翻倍。
6. AAV衣壳的使用趋势:从经典血清型到工程化新型衣壳的加速应用
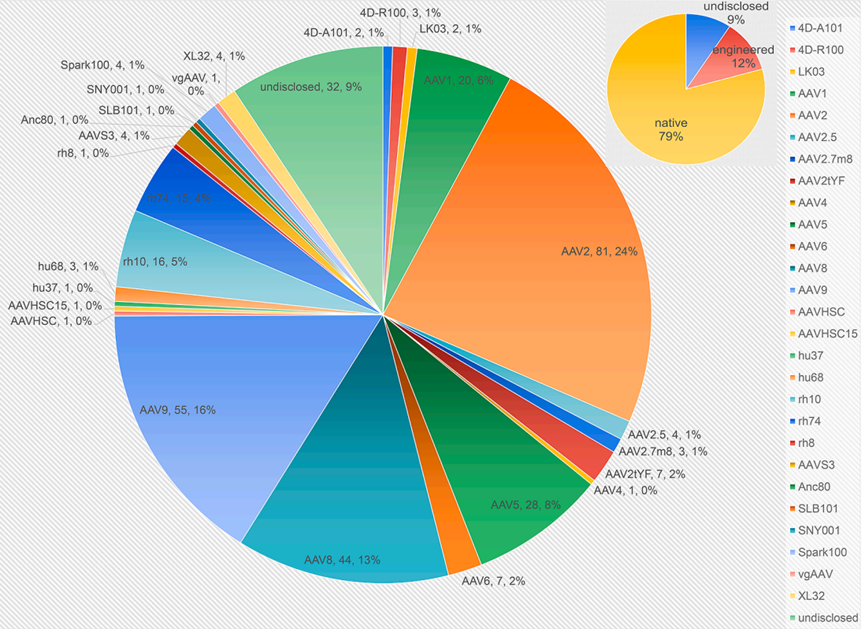
图3 AAV衣壳在340项临床试验中的使用分布情况。图中展示了不同类型AAV衣壳在当前临床试验中的使用比例。
二、AAV基因治疗的现实挑战:临床试验热潮 vs 商业化遇冷
尽管AAV基因治疗在临床试验领域持续扩展,然而“单基因病可治愈”的理想愿景,至今仍难以完全实现。究其原因,不仅限于技术或数据问题,背后也潜藏着多个维度的挑战:
- 在动物模型中,研究往往关注“改善的参数”,却忽略“未改变的指标”,低估了只缓解主要症状无法带来最佳患者获益的现实。
- 对疾病机制与AAV药物生物学的理解仍不够深入。
- 缺乏精准生物标志物、临床试验设计存在偏差,且部分商业预期存在误判,这也影响了融资决策。
1. “肝脏靶向”曾是AAV治疗明星,但如今热度减退
肝脏靶向的基因治疗曾一度被视为AAV基因治疗的“明星领域”,甚至被称作“唾手可得的果实”。业界曾普遍预计,针对血友病A和B的基因治疗产品将成为重磅药物。这两个适应症之所以被视为基因治疗的理想靶点,是因为它们均为隐性单基因疾病,仅需少量凝血因子水平的提升即可显著减少出血事件风险,更具治疗敏感性。同时,这类治疗方案也被认为具有良好的商业化潜力。
然而,尽管已有数据充分证明相关产品在疗效和安全性方面表现优异,行业却在进入后期开发阶段后逐渐将研发重心从血友病转移。而更令人意外的是,尽管已有产品获批,患者和临床医生对这些新疗法的接受度却仍然非常缓慢。
2. “千亿押注”换来“寂寞上线”
过去几年,业界在血友病基因治疗药物的开发上投入了巨额资金,总体投入高达数十亿美元,且多个企业几乎同步开展了平行研发项目。例如,在血友病B方面,参与研发的企业包括辉瑞/Spark、BioMarin、UniQure/CSL、Freeline Therapeutics、百特/武田、以及信念生物/武田中国等;在血友病A方面,则有BioMarin、辉瑞/Sangamo、Spark/罗氏、UCL/St. Jude、拜耳/Ultragenyx以及武田等。
截至目前,针对这些适应症已累计开展超过40项干预性临床试验。基于这些临床研究成果,目前已有3款血友病基因治疗药物获得批准上市:Hemgenix(治疗血友病B),单人治疗费用高达350万美元;Beqvez/Durveqtix(同为血友病B治疗药物),价格同为350万美元;以及Roctavian(治疗血友病A),单次治疗价格为290万美元。
截至目前,Hemgenix 和 Roctavian 的市场接受度仍然偏低,而新近获批的 Beqvez 预计也将面临类似挑战。事实上,Hemgenix 的销售情况之低迷,以至于其生产商 CSL 在财报中甚至未单独披露该产品的销售数据。BioMarin 的 Roctavian 在 2023 年的销售额为 350 万美元,2024 年上半年累计销售额为 700 万美元,远低于其早期预估的 2023 年单年销售额 1.5 亿美元。
值得一提的是,辉瑞的 Beqvez(又名 Durveqtix)虽已于 2024 年中获得批准,但目前尚无销售数据公布。讽刺的是,Beqvez 获批不久后,辉瑞却决定终止其 AAV 平台下的血友病 A 项目,尽管其关键性临床试验数据表现积极。同样,罗氏也在完成 2 期研究后宣布停止其血友病 A 候选药物的研发。在激烈竞争与上市后市场反应冷淡的双重压力下,血友病基因治疗药物未来是否真能成为“重磅炸弹”甚至进入主流治疗路径,仍充满不确定性。
3. 另一种可能的未来:Luxturna的“中庸之路”
美国首个获批的AAV基因治疗药物——Luxturna(由Spark Therapeutics与宾夕法尼亚大学联合开发,用于治疗遗传性视网膜营养不良LCA)似乎正在呈现出一种“中间型”的市场表现。LCA是一种非致命性罕见病,目前除基因治疗外无其他有效治疗手段。Luxturna的研发由Jean Bennett和Albert Maguire两位教授在学术机构内推动,起初完全源于科研热情,后来才吸引到商业资本的关注。该产品获批后,华尔街分析师曾预计其2022年销售额将达到3.46亿美元。然而实际销售远低于这一预期,年销售额区间大致在1000万至5000万美元之间,尽管如此,Luxturna的市场表现仍明显优于Roctavian。
其中一个重要原因可能在于,Luxturna目前是市场上价格最低的AAV基因药物,双眼治疗总价为85万美元。总体来看,除非针对的是真正存在巨大未满足医疗需求的疾病,罕见病AAV基因治疗产品的商业表现更可能落在这一“中间型”区间,而非成为传统意义上的“重磅炸弹”药物。
4. AAV基因治疗的第三种轨迹:是商业奇迹,还是道德困境?
还有一类AAV基因治疗药物呈现出第三种发展轨迹,即面向致死性疾病,但这些疾病已有标准治疗方案可用。这类产品的代表包括Zolgensma(由Avexis/诺华开发)和Elevidys(由Sarepta/罗氏合作开发)。在目前获批的7款AAV基因治疗药物中,仅有这两款实现了超过10亿美元的累计销售额,跻身“重磅炸弹”药物行列——就罕见病药物而言,这一成绩尤为罕见。根据诺华的官方数据,Zolgensma自上市以来,已在全球范围内通过临床试验、扩展用药计划和商业渠道累计治疗超过4000名患者;Sarepta则在JP摩根会议上披露,Elevidys已用于治疗600多名DMD患者,市场渗透率尚不足5%,未来仍具备显著的增长潜力。令人瞩目的是,尽管这两款药物在使用过程中报告了更多不良事件(包括严重不良反应甚至死亡病例),其市场接受速度仍远超血友病类AAV药物。这说明,在致命性疾病治疗中,即便存在风险,患者和临床医生仍倾向于积极尝试AAV疗法,这为该类产品打开了更广阔的商业化前景。
5. 第四类情形:Upstaza的“极罕疾病困境”
Upstaza(又名 Kebilidi)展现了AAV基因治疗药物的另一种独特发展路径。Upstaza由PTC公司开发,用于治疗一种极其罕见且严重的疾病——芳香族氨基酸脱羧酶(AADC)缺乏症。全球患者估计不足350人。该产品定价高达371万美元,在2023年的销售额为1300万美元,预计到2025年销售额可达4000–5000万美元,峰值营收预期为2.66亿美元。不过,这一销售预期颇令人疑惑,因为目前全球约一半AADC患者已通过两个独立的临床项目接受治疗:一项由Krys Bankiewicz博士与哥伦布儿童基金会合作开展,另一项则是PTC自身的开发项目。因此,PTC主要的商业收益很可能已通过其出售“罕见儿科疾病优先审评券(PRV)”而提前兑现,售价为1.5亿美元。从目前AADC治疗所需的生产成本与剂量估算,如果以非盈利方式推进,该病的全球治疗成本仅需约1500万美元;而在当前的商业化定价体系下,这一金额只能治疗4名患儿。
6. AAV商业化的伦理困局:为患者服务,还是为利润服务?
三、AAV临床成功的未来:从实验室到病床
尽管AAV介导的基因治疗在治疗罕见病方面展现出巨大潜力,但在真正实现临床成功的道路上,仍面临诸多挑战。提升安全性与疗效是当前研究和开发工作的重中之重。
目前,AAV介导的基因治疗在罕见病与特发性疾病领域仍面临诸多挑战,提升其安全性与疗效仍是当前最核心的任务之一。最早一项采用AAV2衣壳,通过静脉注射递送凝血因子IX(FIX)的临床试验中,就观察到患者转氨酶水平无症状升高,随后FIX在外周血中的表达水平下降。研究人员由此推测,患者对AAV衣壳产生的免疫反应可能是导致表达下降的主要原因。此后,Nathwani团队的临床试验发现,通过糖皮质激素干预可抑制转氨酶升高,并维持FIX的稳定表达[3]。
1. 优化的风险:密码子≠无限强化
为了在更低剂量下实现治疗效果,研究人员开始对AAV载体进行优化,以提升目标基因的表达效率,尤其是针对分泌型蛋白,这一策略在初期被认为是可行路径。优化方向主要集中在启动子、其他调控元件及编码序列本身。其中,自互补型AAV(scAAV)的开发,为某些小尺寸转基因(如治疗SMA)提供了结构上的解决方案。但需引起高度重视的是,Kattenhorn等人的研究指出,提升表达水平,尤其是通过密码子优化,可能会带来意想不到的不良后果[4]。虽然常被忽略,但已有充分证据表明,蛋白质在翻译过程中的正确折叠往往依赖于某些区域中罕见密码子的存在。例如,分泌蛋白穿越细胞膜的过程中,翻译速度的适当放缓有助于信号识别颗粒(SRP)结合信号肽,并将其引导至内质网膜。一旦将这些罕见密码子替换为高频密码子,可能导致蛋白错误折叠,最终被细胞降解或形成聚集体(包涵体);即使未造成完全失活,也可能对蛋白质结构与翻译后修饰造成干扰,影响其正常功能。
研究显示,某些“无义突变”(即不改变氨基酸序列的沉默突变)也可能对药物反应产生显著影响。例如,MDR1基因中的沉默突变会降低化疗及心血管药物的敏感性,COMP基因中的沉默突变则会影响疼痛感知。早在2009年,《Scientific American》就曾报道,至少有50种与人类疾病相关的沉默突变,其机制可能包括剪接改变、mRNA降解导致表达下调、以及因错误折叠导致蛋白功能异常[5]。在所有潜在机制中,错误折叠蛋白在细胞内的积聚尤其值得关注,因为这会激活“未折叠蛋白反应(UPR)”,并可能引发严重的细胞毒性反应。这提醒我们,在基因治疗中进行表达增强设计时,必须全面考量其在转录、翻译、折叠及功能层面的生物学后果。
2. AAV的毒性事件:稀有但致命
在AAV基因治疗的临床试验中,致死性毒性事件虽然极为罕见,但通常与高剂量静脉注射治疗产品密切相关。迄今为止,所有已知与AAV载体相关的致死性毒性案例已被Duan进行了系统回顾[5]。报告指出,患者可能发生因载体引起的肝功能衰竭、肾功能衰竭、心力衰竭,以及最新报道的肺功能衰竭。在最近一例肺衰竭案例中,研究人员推测为注射CRISPR相关调控载体CRD-TMH-001后引发的细胞因子介导的毛细血管渗漏综合征所致。此外,有8例死亡病例与肝毒性有关:其中4例为接受Zolgensma治疗的DMD患者,使用剂量为1.1×10¹⁴ vg/kg;另4例为接受AT132治疗的X连锁肌管性肌病(XLMTM)患者,剂量范围为5×10¹³至3×10¹⁴ vg/kg。前者可能与AAV衣壳介导的免疫反应有关,后者则被认为与患者原发的肝胆系统基础疾病相关。另有一例患者在接受Zolgensma治疗后死亡,死因为补体激活相关的血栓性微血管病(TMA),并伴随金黄色葡萄球菌感染引发的脓毒症。还有一例DMD患者在接受剂量为2×10¹⁴ vg/kg的PF-06939926治疗后死亡,其死因被归因于由先天性免疫反应诱发的心脏损伤。这些案例提醒我们,在推动AAV基因治疗剂型向更高效、更系统递送方向发展的同时,必须对剂量、安全阈值以及患者基础状况进行更为谨慎的评估。
3. 肝毒性(Hepatic Toxicity)
根据Samelson-Jones和George的综述[6],目前多项AAV基因治疗临床试验中均观察到不良事件(AE)和严重不良事件(SAE)。其中,肝毒性的发生率在不同试验中介于20%至90%之间,其严重程度与AAV载体剂量呈正相关。低剂量情况下(如AAV5、AAV6、AAV‑LK03、SPARK100,剂量区间为4.5×10¹¹–6×10¹³ vg/kg)多为无症状转氨酶升高;中剂量(如AAV9,1.1×10¹⁴ vg/kg)可导致可通过激素治疗逆转的肝功能衰竭;而高剂量(如AAV8,1.3×10¹⁴–3.5×10¹⁴ vg/kg)则可引发不可逆的肝衰竭甚至死亡。不过,文中未充分强调一个关键差异:几乎所有无症状患者为成人,而重症肝毒性多出现在儿童患者中。这一年龄因素可能在风险评估中具有重要意义。
4. 血栓性微血管病(TMA)
血栓性微血管病(TMA)虽属罕见,但在多个使用AAV9衣壳的临床项目中被观察到,涉及适应症包括SMA、不同类型的DMD和Danon病,剂量在1.1×10¹⁴–3×10¹⁴ vg/kg之间。所有病例均通过血液透析和补体抑制剂Eculizumab管理。此外,LogicBio开展的甲基丙二酸血症临床试验中,一名不满2岁的儿童在接受5×10¹³ vg/kg剂量治疗后也出现TMA,经液体补充和静脉营养治疗后好转。在4DMT开展的法布雷病(Fabry)试验中,剂量为1×10¹³ vg/kg时共出现3例非典型溶血性尿毒综合征(TMA的一种形式),其中1例需接受透析治疗。
5. 转基因毒性(Transgene Toxicity)
部分试验数据也引发了对转基因本身毒性的关注。例如,辉瑞在DMD试验中排除了携带特定突变的患者;其在血友病A试验中观察到因转基因过表达而出现血栓事件;Freeline在血友病B试验中也有类似的过表达致血栓现象。
有趣的是,截至目前所有血友病基因治疗试验中,均未报告有ADA(Anti-Drug Antibody)产生,这可能支持“肝脏递送可诱导免疫耐受”这一假设,使患者更容易耐受由AAV表达的蛋白产物。
6. 神经毒性(Neurotoxicity)
此外,在肌萎缩侧索硬化症(ALS)和Batten病的试验中,观察到神经毒性,其中ALS患者通过MRI检测到中枢神经异常信号,DRG(背根神经节)也被认为可能参与其中,部分患者还伴有无症状性转氨酶升高。在Lysogene开展的MPSIIIA试验中,因MRI检查中发现高信号改变,整个试验曾被临时叫停。
四、结语:前路漫漫,仍需奋进
尽管我们仍处于对AAV递送成功机制的早期探索阶段,但社会和经济因素与衣壳工程或启动子优化一样,决定着基因治疗能否真正“从实验室走向临床”。
作为致力于兑现 “从实验台到病床”承诺的研究人员,我们的成就令人瞩目——从1891年发现SMA疾病,到1995年发现SMN1致病基因,再到某新生儿在出生第4天确诊、第7天即接受FDA批准的AAV治疗。如今,全球正在开展的AAV临床试验已超过300项,跨项目、跨机构的临床经验交流(如肝毒性管理)将成为治疗成功的关键。
我们必须正视社会和经济因素现实,重返未来(Back to the Future),以更全面的视角推进AAV基因治疗事业的发展。(未完待续)
关于派真
作为一家专注于AAV 技术十余年,深耕基因治疗领域的CRO&CDMO,派真生物可提供从载体设计、构建到 AAV、慢病毒和 mRNA 服务的一站式解决方案。凭借深厚的技术实力、卓越的运营管理和高标准的服务交付,我们为全球客户提供一站式CMC解决方案,包括从早期概念验证、成药性评估到IIT、IND及BLA的各个阶段。
凭借我们独立知识产权的π-alphaTM 293 细胞AAV高产技术平台,我们能将AAV产量提高多至10倍,每批次产量可达1×10¹⁷vg,以满足多样化的商业化和临床项目需求。此外,我们定制化的mRNA和脂质纳米颗粒(LNP)产品及服务覆盖药物和疫苗开发的各个阶段,从研发到符合GMP的生产,提供端到端的一站式解决方案。
